{kind=link}
This repository contains code for reproducing results in the MethylVI paper.
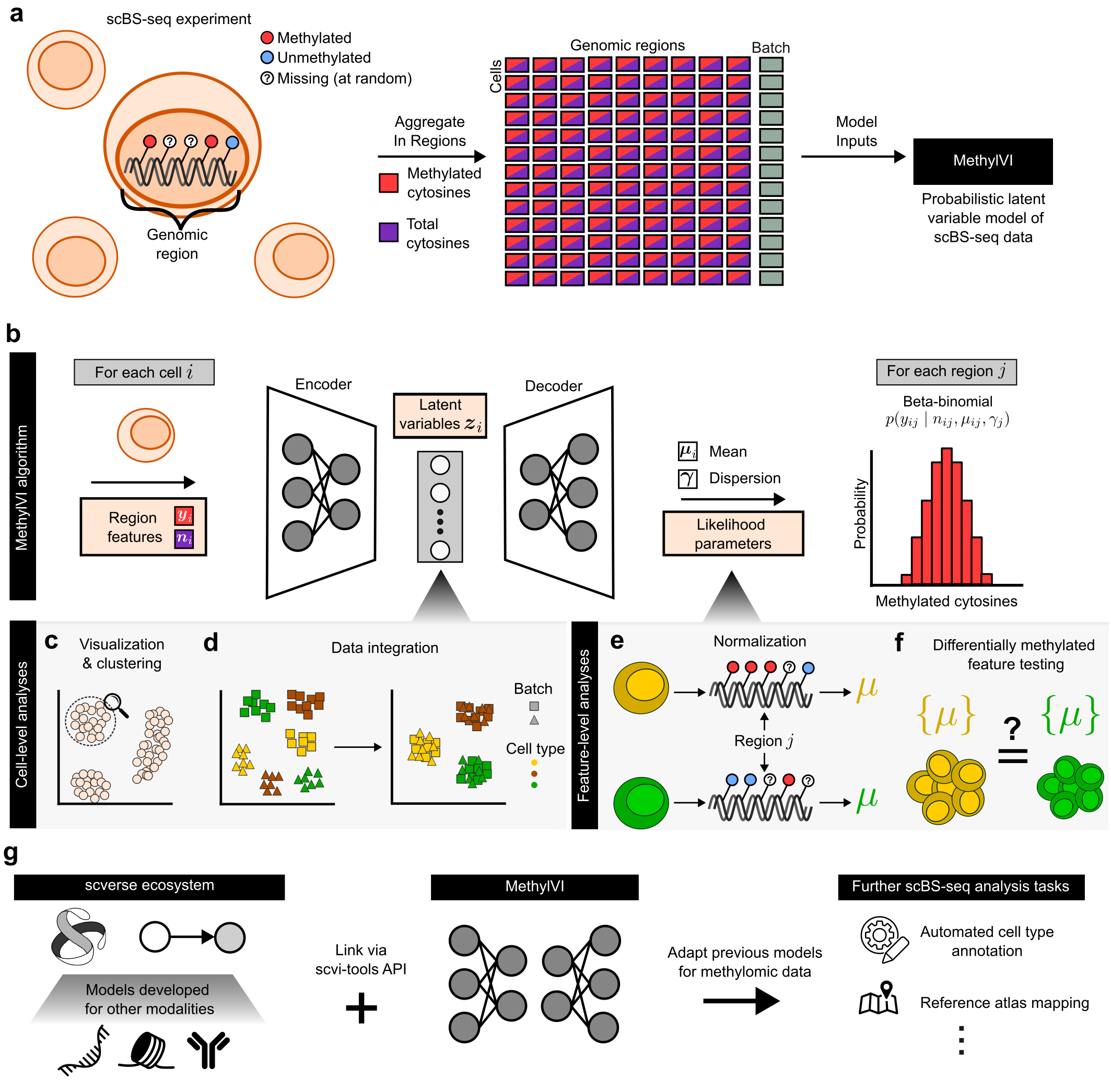
MethylVI is a generative model of single-cell bisulfite sequencing (scBS-seq) data designed to recover latent representations of cells' underlying epigenomic state while controlling for technical sources of variation.
A reference implementation of MethylVI is available in scvi-tools. Google Colab notebooks demonstrating its use are available in the package's accompanying tutorials.
- Produce compressed representations of high-dimensional scBS-seq datasets
- Integrate scBS-seq datasets collected using different experimental conditions (e.g. different BS-seq protocols)
- Perform differentially methylated feature testing
- Map new "query" datasets to previously constructed scBS-seq reference atlases
- And more! MethylVI easily integrates with other scvi-tools/scverse models, and we're excited to see what other use cases emerge.
This software was designed and tested on a machine running Rocky Linux 9.5, with Python 3.10.13,
PyTorch 1.13.1, and CUDA 12.4. For a full list of all external Python package dependences used in this project,
see the Conda environment files methyl-vi-environment.yml and allcools-environment.yml.
When available, this software leverages graphical processing units (GPUs) to accelerate neural network training and evaluation. In our experiments we found that models trained with the aid of a GPU converged in less than 30 minutes (and usually much sooner), with the exact time depending on the size of a given dataset. Systems lacking suitable GPUs may take a longer time to train/evaluate models. Our experiments were conducted using an NVIDIA RTX 2080 TI GPU; other GPUs should also work as long as they have sufficient memory (~2GB).
See notebooks folder for Jupyter notebooks to reproduce individual figures. Within the notebooks
can be found additional information on what datasets must be downloaded/what experiment scripts must
be run to produce the figure.
Due to dependency conflicts between ALLCools, a Python package used to preprocess scBS-seq data in the ALLC file format, and scvi-tools, separate Python environments are required for downloading data vs running experiments. Instructions for setting up these environments can be found below.
- Git clone this repository.
- Create and activate the ALLCools conda environment via
conda env create -f allcools-environment.yml conda activate allcools-env - Navigate to the
datadirectory and run the corresponding Python script for a given dataset.
- Git clone this repository.
- Create and activate the MethylVI conda environment via
conda env create -f methyl-vi-environment.yml conda activate methyl-vi-env - Navigate to the
experiment_scriptsdirectory and run corresponding Python scripts for training models.
If you find contrastiveVI useful for your work, please consider citing our preprint:
TODO: Update once preprint is on bioRxiv.