
This is the variant_calling pipeline from the Sequana projet
| Overview: | Variant calling from FASTQ files |
|---|---|
| Input: | FASTQ files from Illumina Sequencing instrument |
| Output: | VCF and HTML files |
| Status: | production |
| Citation: | Cokelaer et al, (2017), 'Sequana': a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352 |
You can install sequana_variant_calling pipeline using:
pip install sequana_variant_calling --upgrade
I would recommend to setup a sequana_variant_calling conda environment executing:
conda env create -f environment.yml
where the environment.yml can be found in the https://github.com/sequana/variant_calling repository.
Later, you can activate the environment as follows:
conda activate sequana_variant_calling
Note, however, that the recommended method is to use singularity/apptainer as explained here below.
sequana_variant_calling --input-directory DATAPATH --reference-file measles.fa
This creates a directory variant_calling. You just need to move into the directory and execute the script:
cd variant_calling sh variant_calling.sh
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s variant_calling.rules -c config.yaml --cores 4 --stats stats.txt
you can also edit the profile file in .sequana/profile/config.ya,l
Or use sequanix interface.
With singularity, initiate the working directory as follows:
sequana_variant_calling --use-singularity --singularity-prefix ~/.sequana/apptainers
Images are downloaded in a global direcory (here .sequana/apptainers) so that you can reuse them later.
and then as before:
cd variant_calling sh variant_calling.sh
if you decide to use snakemake manually, do not forget to add singularity options:
snakemake -s variant_calling.rules -c config.yaml --cores 4 --stats stats.txt --use-singularity --singularity-prefix ~/.sequana/apptainers --singularity-args "-B /home:/home"
If you rely on singularity/apptainer, no extra dependencies are required (expect python and https://damona.readthedocs.io). If you cannot use apptainer, you will need to install some software:
- bwa
- freebayes
- picard (picard-tools)
- sambamba
- minimap2
- samtools
- snpEff you will need 5.0 or 5.1d (note the d); 5.1 does not work.
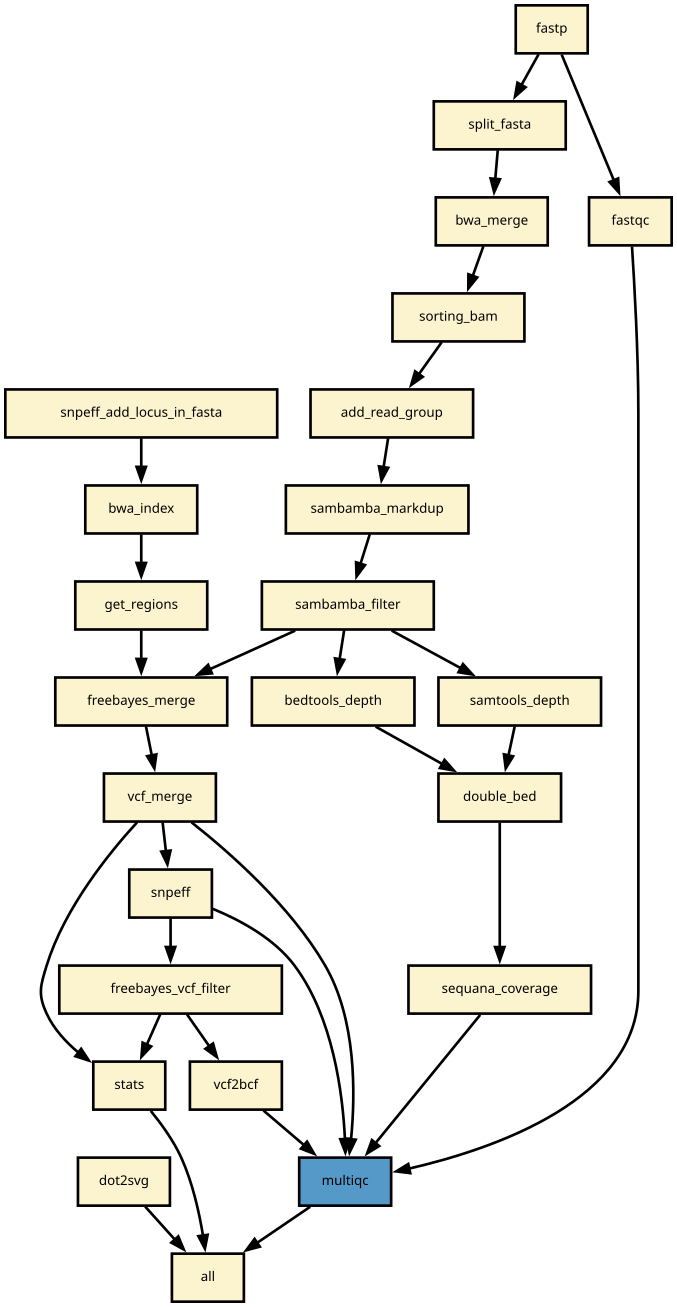
Snakemake variant calling pipeline is based on tutorial written by Erik Garrison. Input reads (paired or single) are mapped using bwa and sorted with sambamba-sort. PCR duplicates are marked with sambamba-markdup. Freebayes is used to detect SNPs and short INDELs. The INDEL realignment and base quality recalibration are not necessary with Freebayes. For more information, please refer to a post by Brad Chapman on minimal BAM preprocessing methods.
The pipeline provides an analysis of the mapping coverage using sequana coverage. It detects and characterises automatically low and high genome coverage regions.
Detected variants are annotated with SnpEff if a GenBank file is provided. The pipeline does the database building automatically. Although most of the species should be handled automatically, some special cases such as particular codon table will required edition of the snpeff configuration file.
Finally, joint calling is also available and can be switch on if desired.
Let us download an ecoli reference genome and the data set used to create the assembly. All tools used here below can be installed with damona (or your favorite environment manager):
pip install damona damona create TEST damona activate TEST damona install pigz damona install sratoolkit # for fasterq-dump damona install datasets
Then, download the data:
fasterq-dump SRR13921546 pigz SRR*fastq
and the reference genome with its annnotation:
datasets download genome accession GCF_000005845.2 --include gff3,rna,cds,protein,genome,seq-report,gbff unzip ncbi_dataset.zip ln -s ncbi_dataset/data/GCF_000005845.2/GCF_000005845.2_ASM584v2_genomic.fna ecoli.fa ln -s ncbi_dataset/data/GCF_000005845.2/genomic.gff ecoli.gff
Initiate the pipeline:
sequana_variant_calling --input-directory . --reference-file ecoli.fa --aligner-choice bwa_split \
--do-coverage --annotation-file ecoli.gff \
--use-apptainer --apptainer-prefix ~/.sequana/apptainers \
--input-readtag "_[12]."
Explication:
- we use apptainer/singularity
- we use the reference genome ecoli.fa (--reference-file) and its annotation for SNPeff (--annotation-file)
- we use the sequana_coverage tool (True by default) to get coverage plots.
- we use --input-directory to indicatre where to find the input files
- This data set is paired. In NGS, it is common to have _R1_ and _R2_ tags to differentiate the 2 files. Here the tag
are _1 and _2. In sequana we define the a wildcard for the read tag. So here we tell the software that thex ecpted tag follow this pattern: "_[12]." and everything is then automatic.
Then follow the instructions (prepare and execute the pipeline).
You should end up with a summary.hml report.
You can browse the different samples (only one in this example) and get a table with variant calls:
https://raw.githubusercontent.com/sequana/variant_calling/refs/heads/main/doc/table.png
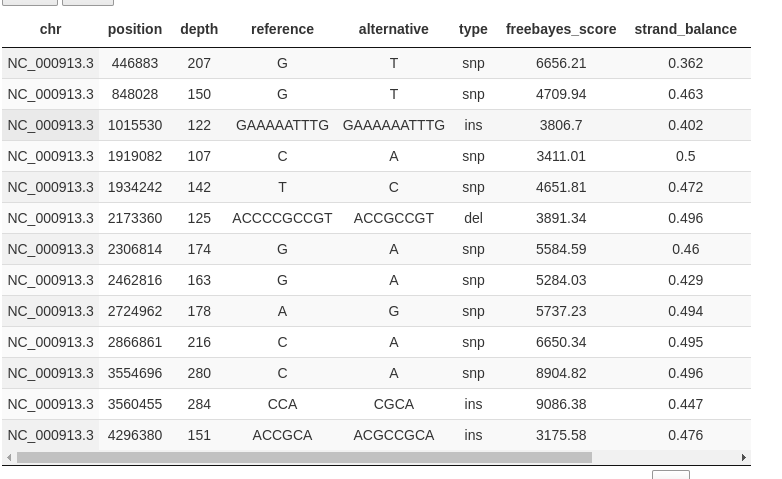{kind=link}
If you set the coverage one, (not recommended for eukaryotes), you should see this kind of plots:
https://raw.githubusercontent.com/sequana/variant_calling/refs/heads/main/doc/coverage.png
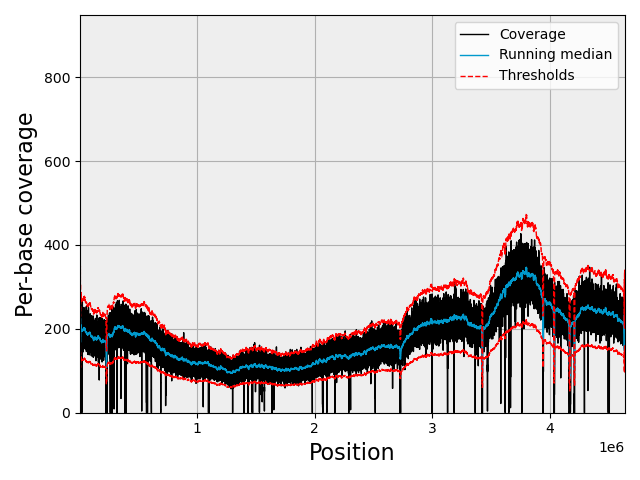{kind=link}
| Version | Description |
|---|---|
| 1.3.0 |
|
| 1.2.0 |
|
| 1.1.2 |
|
| 1.1.1 |
|
| 1.1.0 |
|
| 1.0.2 |
|
| 1.0.1 |
|
| 1.0.0 |
|
| 0.12.0 |
|
| 0.11.0 |
|
| 0.10.0 |
|
| 0.9.10 |
|
| 0.9.5 |
|
| 0.9.4 |
|
| 0.9.3 |
|
| 0.9.2 |
|
| 0.9.1 |
|
| 0.9.0 | First release |
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.