ngsDist is a program to estimate pairwise genetic distances directly, taking the uncertainty of genotype's assignation into account. It does so by avoiding genotype calling and using genotype likelihoods or posterior probabilities.
ngsDist was published in 2015 at Biological Journal of the Linnean Society, so please cite it if you use it in your work:
Vieira FG, Lassalle F, Korneliussen TS, Fumagalli M
Improving the estimation of genetic distances from Next-Generation Sequencing data
Biological Journal of the Linnean Society (2015) 117(1):139-149
ngsDist can be easily installed but has some external dependencies:
- Mandatory:
gcc: >= 4.9.2 tested on Debian 7.8 (wheezy)zlib: v1.2.7 tested on Debian 7.8 (wheezy)gsl: v1.15 tested on Debian 7.8 (wheezy)
- Optional (only needed for testing or auxilliary scripts):
md5sum
To install the entire package just download the source code:
% git clone https://github.com/fgvieira/ngsDist.git
and run:
% cd ngsDist
% make
To run the tests (only if installed through ngsTools):
% make test
Executables are built into the main directory. If you wish to clean all binaries and intermediate files:
% make clean
% ./ngsDist [options] --geno /path/to/input/file --n_ind INT --n_sites INT --out /path/to/output/file
--geno FILE: input file with genotypes, genotype likelihoods or genotype posterior probabilities.--probs: is the input genotype probabilities (likelihoods or posteriors)?--log_scale: Is the input in log-scale?.--n_ind INT: sample size (number of individuals).--n_sites INT: number of sites in input file.--tot_sites INT: total number of sites in dataset.--labels(H) FILE: labels, one per line, of the input sequences;--labelsHassumes there is a header.--pos(H) FILE: position info (chr, pos, allele1, allele2), one per line, of the input sites;--posHassumes there is a header (required for more complex evolutionary models).--call_geno: call genotypes before running analyses.--N_thresh DOUBLE: minimum threshold to consider site; missing data if otherwise (assumes -call_geno)--call_thresh DOUBLE: minimum threshold to call genotype; left as is if otherwise (assumes -call_geno)--pairwise_del: pairwise deletion of missing data.--avg_nuc_dist: use average number of nucleotide differences as distance (by default,ngsDistuses genotype distances based on allele frequency differences). Only pairs of heterozygous positions are actually affected when using this option, with their distance being 0.5 (instead of 0 by default).--evol_model: DNA evolutionary (0) raw p-distance, (1) log-transformed p-distance, (2) JC69, (3) K80, (4) F81, (5) HKY85, (6) TN93. [1]--indep_geno: assume independence between genotypes? If so, skip EM step and just multiply probabilities (faster).--n_boot_rep INT: number of bootstrap replicates [0].--boot_block_size INT: block size (in alignment positions) for bootstrapping. [1]--out FILE: output file name.--n_threads INT: number of threads to use. [1]--verbose INT: selects verbosity level. [1]--seed INT: random number generator seed (only for the bootstrap analysis).
As input, ngsDist accepts both genotypes, genotype likelihoods (GL) or genotype posterior probabilities (GP). Genotypes must be input as gziped TSV with one row per site and one column per individual 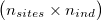
ngsDist accepts both gzipd TSV and binary formats, but with 3 columns per individual 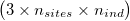
If your data is in VCF format, you can convert it to BEAGLE using bcftools (to filter VCF for [e.g.] biallelic SNPs on autossomal chromosomes that PASS all filters) and plink (to convert to almost the correct format):
bcftools view -f PASS --types snps -m1 -M2 -r `seq -s, 1 22` INPUT.vcf.gz | plink --double-id --vcf /dev/stdin --recode A-transpose --out /tmp/plink
tail -n +2 /tmp/plink.traw | cut -f 1,4- | perl -p -e 's/\bNA\b/-1/g' | gzip > INPUT.geno.gz
# Labels file
zgrep CHROM INPUT.vcf.gz | cut -f 10- | tr "\t" "\n" > INPUT.labels
ngsDist strength is the possibility of taking genotype uncertainty (from genotype likelihoods) into account but, at the moment, it can only calculate simple "p-distance" and JC69 corrected distances. More complex evolutionary models (e.g. K2P, HKY85) are under development.
If you want branch support values on your tree, you can use ngsDist with the option --n_boot_rep and --boot_block_size to bootstrap the input data. ngsDist will output one distance matrix (the first) for the input full dataset, plus --n_boot_rep matrices for each of the bootstrap replicates. After, infer a tree for each of the matrices using the program of your choice and plot them. For example, using FastME on a dataset with 5 bootstrap replicates:
fastme -T 20 -i testA_8B.dist -s -D 6 -o testA_8B.nwk
split the input dataset tree from the bootstraped ones:
head -n 1 testA_8B.nwk > testA_8B.main.nwk
tail -n +2 testA_8B.nwk | awk 'NF' > testA_8B.boot.nwk
and, to place supports on the main tree, use RAxML:
raxmlHPC -f b -t testA_8B.main.nwk -z testA_8B.boot.nwk -m GTRCAT -n testA_8B
or RAxML-NG:
raxml-ng --support --tree testA_8B.main.nwk --bs-trees testA_8B.boot.nwk --prefix testA_8B
The thread pool implementation was adapted from Mathias Brossard's and is freely available from: https://github.com/mbrossard/threadpool