


Alternatively Spliced Pseudo Repeat IN-Gene
ASPRING is a computational tool for detecting Alternative Splicing Repetitive Units (ASRUs) on a gene. It analyzes the outputs of ThorAxe, which provides Multiple Sequence Alignments of exonic regions and generates information about their use on alternative isoforms. You can run aspring through the command line to find duplication events of such exonic regions. This tool will provide information on the duplicated regions through a couple of tables.
Note
ASPRING requires ThorAxe outputs for a single query gene to run. If you don't
have ThorAxe outputs, you can visit the ThorAxe documentation to learn how to install
and run ThorAxe on your data. ThorAxe can also be run using the Ases web server,
which provides a user-friendly interface for running ThorAxe online. Note that you can
skip the PhyloSofS step of Ases to obtain results more quickly for use with aspring.
Once you have ThorAxe outputs, you can use aspring to identify ASRUs for your query
gene.
ASPRING is a Python package that can be installed from PyPI using the pip package
manager. Its pipeline uses R and the HH-suite3. In particular, Rscript from R
and hhmake and hhalign from HH-suite3 must be in the PATH.
You can see the installation instructions for both in the following links:
If you have miniconda installed, you can use it to install both HH-suite3 and R. For example:
conda install -c conda-forge -c bioconda hhsuite conda install -c conda-forge
r-base=4.2.2Also, you will need to know the path where the HH-suite3 scripts are installed, as
ASPRING needs to access reformat.pl. If you installed HH-suite3 using the base
environment miniconda on a Linux, then the path will be something like
~/miniconda3/scripts.
The ASPRING package ships with a renv environment (located at src/aspring/R_scripts)
that will be automatically executed, so you do not need to worry about installing the R
dependencies.
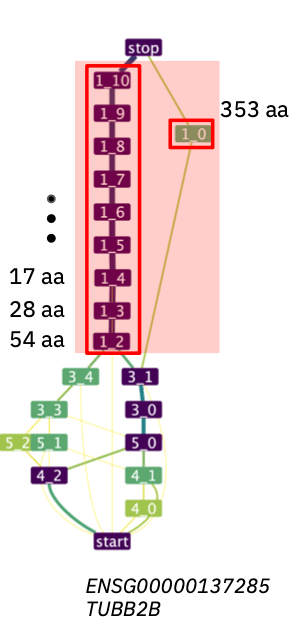
The figure shows an example of an Alternatively Spliced Repetitive Unit (ASRU) composed by two spliced-repeats (s-repeats), one of them defined by a single s-exon and the other one comprising multiple s-exons.
The nodes are the s-exons. The opaque red boxes are the s-repeats, and the transparent red box is the ASRU.
The s-repeats are repetitive units identified by ASPRING consisting of one or more s-exons alternatively included or excluded in different isoforms. Note that s-repeats are called instances on the output tables.
aspring is a Python-based command-line tool that helps identify Alternative Splicing
Repetitive Units (ASRUs) from Thoraxe outputs for a single query gene. The tool executes
several steps that involve converting data, creating HMM profiles, aligning profiles,
parsing and filtering alignments, and generating ASRUs and spliced-repeats (s-repeats) tables for the query gene.
Here is how to run the script after installing the package:
Open your terminal.
Run the script with the following command:
aspring --gene GENE_NAME --path_data PATH_TO_THORAXE_OUTPUTS --path_hhsuite_scripts PATH_TO_HHSUITE_SCRIPTS
To query a specific gene, replace
GENE_NAMEwith the corresponding Ensemble ID. If ThorAxe outputs are in the current working directory,--path_dataparameter can be avoided. In case thereformat.plscript from the HH-suite3 is in the path indicated by theHHSUITE_SCRIPTSenvironment variable,--path_hhsuite_scriptsparameter can be omitted. Otherwise, replacePATH_TO_HHSUITE_SCRIPTSwith the path to thereformat.plscript directory. ReplacePATH_TO_THORAXE_OUTPUTSwith the path to the ThorAxe outputs directory for the gene of interest.Optional arguments are available to customize the behavior of the script. Run the command
aspring --helpto see the full list of options.
The script will execute several steps and generate output files containing ASRU and s-repeats tables for the query gene.
To ease the use and installation of ASPRING, we have created a Docker image that
you can easily download and run. The aspring Docker image is available on
Docker Hub. To run the aspring tool using the Docker image, you must have
Docker installed on your system. You can download and install Docker from the
official website. Once Docker is installed, you can run aspring using the
following command:
sudo docker run --mount type=bind,source=$(pwd),target=/data diegozea/aspring aspring --gene GENE_NAMEIn this command, we use the docker run command to run aspring. We are
mounting the current working directory using the --mount option, which is
necessary for providing access to the data files required by aspring. The
--mount option takes two parameters: type and source. type specifies
the type of mount to use. In this case, we use a bind mount, which allows us
to mount a directory from the host system to the container; that is a
requirement to enable aspring to see the input files and to let it save
the output files in your filesystem. source specifies the source directory to
mount. In this case, we use $(pwd) to select the current working directory as
the source. We are also specifying the target directory as /data in the
container. This means that the files from the current working directory on the
host system will be available in the /data directory in the container.
The aspring tool requires R and the HH-suite3, which are already
installed in the Docker image. Therefore, there is no need to specify
--path_hhsuite_scripts or --path_data; the last one is set to /data by
default.
Finally, we specify the --gene option with GENE_NAME to run aspring on that gene.
ASPRING is a tool for detecting Alternative Splicing Repetitive Units (ASRUs) on a gene. The
pipeline consists of nine steps, each of which can be executed separately, but it is
recommended to run the main script aspring to execute the entire pipeline. Only steps 1,
2, and 3 require HH-suite3 and step 6 requires R. You can use the -h argument to
show the arguments for each step.
The pipeline steps are:
step_01_preprocess: Reformat s-exons fasta files to a2m.step_02_hmm_maker: Generates a Hidden Markov Model (HMM) profile for each s-exon.step_03_hmm_aligner: HMM-HMM alignment of all the s-exons combinations.step_04_gettable: Parses the alignment files and creates a table.step_05_filter: Filter the table to keep gene duplication pairs based on identity, coverage, p-value and number of species in the MSAs.step_06_stats: Generates statistics on the filtered duplicated regions.step_07_reformat: Reformat the previous outputs to add the information about the duplicated regions.step_08_ASRUs: Identifies the Alternative Splicing Repetitive Units (ASRUs) on the gene.step_09_clean: Removes the intermediate files generated during the pipeline.step_10_struct: Maps s-exons and ASRUs to the protein structures obtained from AlphaFold DB.
Note that the main script aspring runs the entire pipeline automatically. However,
the user can also execute the scripts of each pipeline step individually for more control
over the pipeline.
For a given gene (Ensembl Gene ID), ASPRING returns:
{gene}_ASRUs_table.csv{gene}_instances_table.csv{gene}_duplication_pairs.csv{gene}_eventsDup_withCols.txtDupRaw/{gene}folder containing thes-exon_A.s-exon_B.hhrfiles (HMM-HMM alignments)structuresfolder containing the isoform structures obtained from AlphaFold DB (.pdbfiles) and a set of PyMol scripts (.pmlfiles) with the s-exons and ASRUs selected and colored in the structure.
This table provides information on the Alternatively Spliced Repeat Units (ASRUs) detected
for the given gene. Each row corresponds to a distinct ASRU and provides the following
information:
gene: The Ensembl Gene ID for the given gene.ASRU: The set s-repeats belonging to the ASRU.Nbinstances: The number of Alternatively Spliced Pseudo Repeats of the ASRU that were found in the exonic regions of the gene.max: The length of the longest s-repeat of the ASRU, in residues.min: The length of the shortest s-repeat of the ASRU, in residues.moy: The mean length of the instances of the ASRU, in amino acid residues.median: The median length of the instances of the ASRU, in residues.std: The standard deviation of the lengths of the instances of the ASRU, in amino acid residues.eventsRank: The rank/position of the alternative splicing events involving the ASRU in theases.csvoutput table from ThorAxe — from the most to the least conserved/frequent.
This table provides information on the instances of ASRUs detected for the given gene.
Each row corresponds to a distinct instance and provides the following information:
instance: The sequence of the s-repeat, in the form of a string of amino acid residues.size: The length of the s-repeat, in amino acid residues.NbSex: The number of exonic regions where the s-repeat was detected.ASRU: The set of homologous/duplicated s-exons that belong to the ASRU to which the s-repeat- belongs.
gene: The Ensembl Gene ID for the given gene.
This table provides information on the pairs of exonic regions that were involved in the duplication events. Each row corresponds to a distinct pair of s-exons and provides the following information:
S_exon_Q: The identifier of the first s-exon.S_exon_T: The identifier of the second s-exon.Gene: The Ensembl Gene ID for the given gene.Prob: The probability score of the alignment of the exonic region pair.E-value: The E-value associated with the alignment of the exonic region pair.P-value: The P-value associated with the alignment of the exonic region pair.Score: The alignment score of the alignment of the exonic region pair.Cols_Q: The alignment columns corresponding to the first s-exon, in the format "start-end".Cols_T: The alignment columns corresponding to the second s-exon, in the format "start-end".Length_Q: The length of the first s-exon, in amino acid residues.Length_T: The length of the second s-exon, in amino acid residues.Identities: The percentage of identical residues in the alignment of the exonic region pair.IdCons: The percentage of conserved residues in the alignment of the exonic region pair.Similarity: The fraction of similar residues in the alignment of the exonic region pair.NoSpecies_Q: The number of species in which the first s-exon is conserved.NoSpecies_T: The number of species in which the second s-exon is conserved.
This table provides detailed information on the alternative splicing events in with the ASRUs are involved. Each row corresponds to a distinct event and provides the following information:
gene: The Ensembl Gene ID for the given gene.sexA: The index of the first s-exon in the ASRU.sexB: The index of the second s-exon in the ASRU.rank: The rank of the alternative splicing event, as ordered in the ThorAxe output table from the most to the least conserved/frequent.type: The type of the alternative splicing events, e.g "alternative".statusA: The status of the path with the first s-exon, which can be alternative or canonical.statusB: The status of the path with the first s-exon, which can be alternative or canonical.lePathA: Number of s-exons in the path with the first s-exon.lePathB: Number of s-exons in the path with the second s-exon.exclu: A boolean indicating whether the event involves mutually exclusive s-exons.pval: The P-value associated with the alignment of the exonic region pair.ncols: The number of columns in the alignment.leA: The length of the first s-exon, in amino acid residues.leB: The length of the second s-exon, in amino acid residues.typePair: The type of the alternative splicing event.ColA: The alignment columns corresponding to the first s-exon, in the format "start-end".ColB: The alignment columns corresponding to the second s-exon, in the format "start-end".
This directory contains the isoform structures predicted by AlphaFold 2 that were obtained
from AlphaFold DB. These are the files with the .pdb extension. The folder also contains
a PyMol script for each transcript whose isoform has a known downloaded structure. Those
scripts have a name composed of the Ensembl gene name, followed by the transcript name, and ending with
the file name of the AlphaFold structure and the .pml extension. For example,
ENSG00000007866_ENST00000338863_AF-Q99594-F1-model_v4.pml is the PyMol script for the isoform
corresponding to the transcript ENST00000338863 of the gene ENSG00000007866.
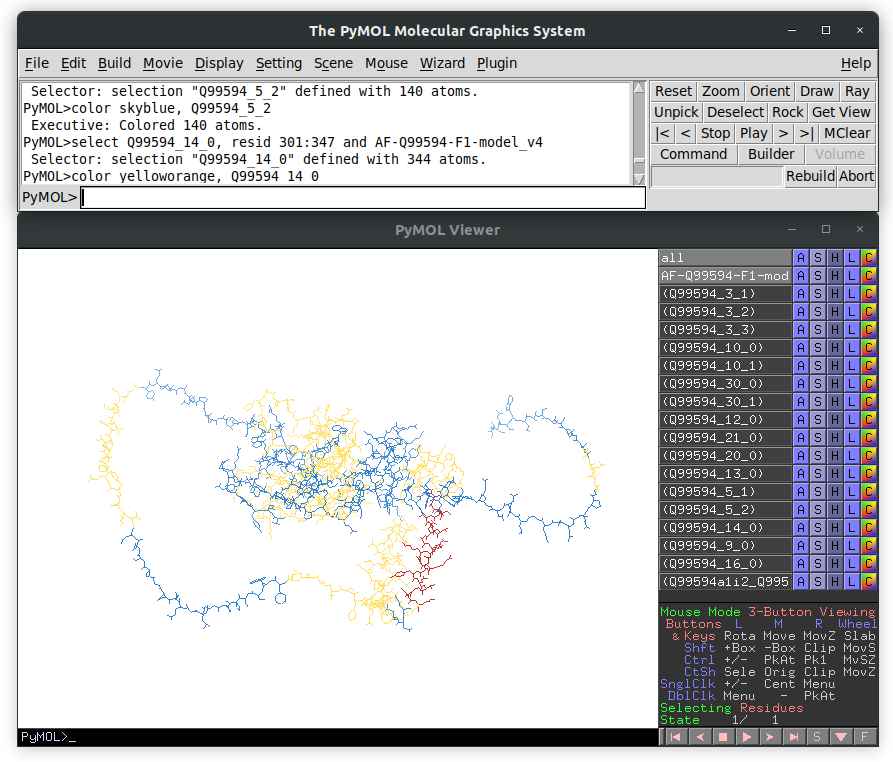
When you run that script using PyMol as:
pymol ENSG00000007866_ENST00000338863_AF-Q99594-F1-model_v4.pmlYou will see the AlphaFold predicted structure of the isoform with the s-exon colored in
alternating light blue and yellow, and the ASRUs in red and green. For example, the
following image shows the PyMOL session for the ENST00000338863 transcript:
This project has been set up using PyScaffold 4.4. For details and usage information on PyScaffold see https://pyscaffold.org/.