Haplotype inference and phasing for Short Tandem Repeats
![]()
Author: Thomas Willems [email protected]
License: GNU v2
Introduction
Requirements
Installation
Quick Start
Tutorial
In-depth Usage
Data Requirements
Phasing
Speed
Default Filtering
Call Filtering
Additional Usage Options
File Formats
FAQ
Help
Citation
Short tandem repeats (STRs) are highly repetitive genomic sequences comprised of repeated copies of an underlying motif. Prevalent in most organisms' genomes, STRs are of particular interest because they mutate much more rapidly than most other genomic elements. As a result, they're extremely informative for genomic identification, ancestry inference and genealogy.
Despite their utility, STRs are particularly difficult to genotype. The repetitive sequence responsible for their high mutability also results in frequent alignment errors that can complicate and bias downstream analyses. In addition, PCR stutter errors often result in reads that contain additional or fewer repeat copies than the true underlying genotype.
HipSTR was specifically developed to deal with these errors in the hopes of obtaining more robust STR genotypes. In particular, it accomplishes this by:
- Learning locus-specific PCR stutter models using an EM algorithm
- Mining candidate STR alleles from population-scale sequencing data
- Employing a specialized hidden Markov model to align reads to candidate alleles while accounting for STR artifacts
- Utilizing phased SNP haplotypes to genotype and phase STRs
In our opinion, all of these factors make HipSTR the most reliable tool for genotyping STRs from Illumina sequencing data.
To compile HipSTR, the following packages are required:
- make
- g++
- zlib
- libhts
- libbz2
- liblzma
If you are running Ubuntu 16+, they can be easily installed by running:
apt install make g++ zlib1g-dev libhts-dev libbz2-dev liblzma-dev
HipSTR requires a standard c++ compiler. To obtain HipSTR, use:
git clone https://github.com/HipSTR-Tool/HipSTR
To build, use Make:
cd HipSTR
make
The commands will construct an executable file called HipSTR in the current directory, for which you can view detailed help information by typing
./HipSTR --help
To run HipSTR in its most broadly applicable mode, run it on all samples concurrently using the syntax:
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--str-vcf str_calls.vcf.gz
- bams : a comma-separated list of BAM/CRAM files generated by BWA-MEM and sorted and indexed using samtools
- regions : a BED file containing the coordinates for each STR region of interest. Download BED files for various organisms, including humans, from here
- fasta : FASTA file containing the sequence for each chromosome in the BED file. This build's coordinates must match those of the STR regions
- str-vcf : The output path for the STR genotypes
For each region in str_regions.bed, HipSTR will:
- Learn a stutter model for each locus
- Use the stutter model and haplotype-based alignment algorithm to genotype each individual
- Output the resulting STR genotypes to str_calls.vcf.gz, a bgzipped VCF file. This VCF will contain calls for each sample in any of the BAM/CRAM files' read groups.
To demonstrate how you can quickly apply HipSTR to whole-genome sequencing datasets, we've built a simple tutorial. In less than 10 minutes, this tutorial will teach you how to genotype ~600 STRs in a deeply sequenced trio of individuals and inspect the results.
HipSTR has a variety of usage options designed to accomodate scenarios in which the sequencing data varies in terms of the number of samples and the coverage. Most scenarios will fall into one of the following categories:
- 100 or more low-coverage (~5x) samples
- Sufficient reads for stutter estimation
- Sufficient reads to detect candidate STR alleles
- Use de novo stutter estimation + STR calling with de novo allele generation
- 20 or more high-coverage (~30x) samples
- Sufficient reads for stutter estimation
- Sufficient reads to detect candidate STR alleles
- Use de novo stutter estimation + STR calling with de novo allele generation
- Handful of low-coverage (~5x) samples
- Insufficient reads for stutter estimation
- Insufficient reads to detect candidate STR alleles
- Use external/default stutter models + STR calling with a reference panel
- Handful of high-coverage (~30x) samples
- Insufficient samples for stutter estimation
- Sufficient reads to detect candidate STR alleles
- Use external/default stutter models + STR calling with de novo allele generation
This mode is identical to the one suggested in the Quick Start section as it suits most applications. HipSTR will output the STR genotypes in bgzipped VCF format to str_calls.vcf.gz
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--str-vcf str_calls.vcf.gz
The sole difference in this mode is that we no longer learn stutter models using the EM algorithm but instead input them from the stutter-in file. For more details on the stutter model file format, see below.
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--stutter-in ext_stutter_models.txt
--str-vcf str_calls.vcf.gz
If you don't have access to external stutter models for the stutter-in option, use def-stutter-model. This will use a simplistic stutter model for all loci (see the HipSTR help message for specifics).
This mode is very similar to mode 2, except that we provide an additional VCF file containing known STR genotypes at each locus using the str-vcf option. HipSTR will not identify any additional candidate STR alleles in the BAMs/CRAMs when this option is specified, so it's best to use a VCF that contains STR genotypes for a wide range of populations and individuals.
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--stutter-in ext_stutter_models.txt
--ref-vcf ref_strs.vcf.gz
--str-vcf str_calls.vcf.gz
If you don't have access to external stutter models for the stutter-in option, use def-stutter-model. This will use a simplistic stutter model for all loci (see the HipSTR help message for specifics).
To genotype STRs, HipSTR requires Illumina sequencing data. However, as the depth of sequencing and the read length in these datasets can vary dramatically, here we briefly describe key factors to consider before generating data for HipSTR analyses.
Because of the repetitive nature of STRs, reads that do not fully extend across the repeat only provide a lower bound on its length. While this lower bound is informative and is leveraged by HipSTR, obtaining accurate and robust STR genotypes requires reads that fully extend across the repetitive sequence (i.e. spanning reads). The number of reads that span an STR is a function of the read length, the sequencing depth, and the length of the repeat (as well as various other factors). The interplay between these factors is relatively complex, but Figure 2 in a recent review by Press et al. nicely highlights these dependencies. As one would intuitively expect, using longer read lengths and higher sequencing coverage increases the number of spanning reads. Conversely, increasing the length of the repeat reduces the number of spanning reads, making it more difficult to accurately genotype long STRs. When the number of spanning reads approaches single digits, you statistically run the risk of observing reads from only 1 out of 2 chromosome copies, making it impossible to correctly call both alleles in a heterozygous individual.
In our own analyses, we've found that 100 bp Illumina reads are sufficient to characterize the majority of STRs in the human genome. However, genotyping STRs that exceed 70bp (such as very long forensic STRs) invariably requires longer reads, but most human STRs are much shorter than this threshold. This read length will likely be sufficient for most model organisms unless their repeats are substantially longer than those in humans.
The optimal minimum sequencing depth for HipSTR largely depends on your intended analyses. If you are interested in studying how STRs mutate or want to identify de novo mutations, 30x coverage is an ideal minimum that allows HipSTR to provide the required high degree of specificity. Conversely, if you are merely interested in studying the allele frequencies for various STRs in a population, 10x coverage will likely be sufficient. However, in this setting, there will likely be many genotyping errors in which heterozygous genotypes are miscalled as homozygotes.
Based on the nature of your sequencing data, one important HipSTR option to consider is min-reads. HipSTR uses the value of this parameter to skip any STRs where few than N reads are available for genotyping across all individuals. By default, this value is 100, as we've found that this is a good minimum threshold for learning stutter models prior to genotyping. If you're analyzing very few samples (e.g. a single mother-father-child trio), you may want to consider lowering this threshold as you will seldom have 100 reads. In this setting, it makes sense to use options like --min-reads 15 --def-stutter-model, where the latter option uses a default stutter model as too few reads are available for accurately inferring one. However, if you're analyzing many samples (e.g. more than ten 30x genomes or more than thirty 10x genomes), it likely doesn't make sense to change this parameter. In these settings, regions with fewer than 100 reads may have high GC content that is problematic for Illumina sequencing, may be difficult to map to, or may merely be too long for your chosen read length.
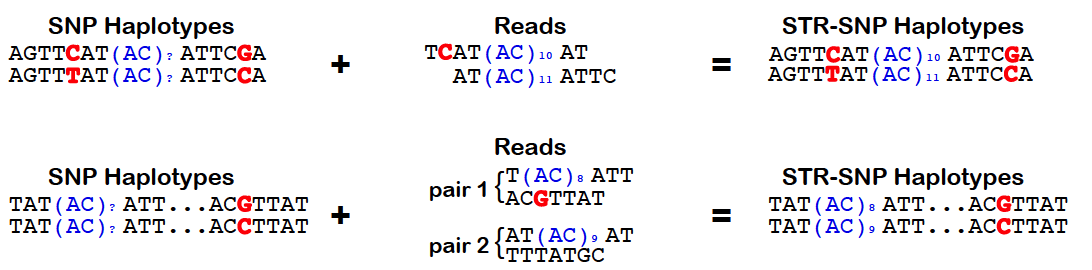
HipSTR utilizes phased SNP haplotypes to phase the resulting STR genotypes. To do so, it looks for pairs of reads in which the STR-containing read or its mate pair overlap a samples's heterozygous SNP. In these instances, the quality score for the overlapping base can be used to determine the likelihood that the read came from each haplotype. Alternatively, when this information is not available, we assign the read an equal likelihood of coming from either strand. These likelihoods are incorporated into the HipSTR genotyping model which outputs phased genotypes. The quality of a phasing is reflected in the PQ FORMAT field, which provides the posterior probability of each sample's phased genotype. For homozygous genotypes, this value will always equal the Q FORMAT field as phasing is irrelevant. However, for heterozygous genotypes, if PQ ~ Q, it indicates that one of the two phasings is much more favorable. Alterneatively, if none of a sample's reads overlap heterozygous SNPs, both phasings will be equally probable and PQ ~ Q/2. To enable the use of physical phasing, supply HipSTR with the snp-vcf option and a SNP VCF containing phased haplotypes. The schematic below outlines the concepts underlying HipSTR's physical phasing model:
HipSTR doesn't currently have multi-threaded support, but there are several options available to accelerate analyses:
- Analyze each chromosome in parallel using the --chrom option. For example, --chrom chr2 will only genotype BED regions on chr2
- Split your BED file into N files and analyze each of the N files in parallel. This allows you to parallelize analyses in a manner similar to option 1 but can be used for increased speed if N is much greater than the number of chromosomes.
HipSTR sometimes automatically filters genotypes on a per-sample basis and will report a missing value in the VCF file. These filters are applied when a sample's data suggests that HipSTR will not be able to produce a reliable genotype. For each locus, a summary of the number of filtered samples is output in the log file. If you specify the --output-filters command line option, a FORMAT field called FILTER will be reported in the VCF for each sample, where PASS designates ok samples and other values indicate the reason for filtering.
Samples with a PASS value should still undergo additional variant filtering (see below), as this merely indicates that no catastrophic issues were encountered during the genotyping process. The table below summarizes the potential filtering reasons:
| Filter | Explanation |
|---|---|
| NO_READS | No alignments were available for the sample at the current STR. If reads overlap the STR in the BAM/CRAM, they may have been filtered due to read quality issues, mapping uniqueness or other reasons |
| FLANK_ASSEMBLY_CYCLIC | During the genotyping process, HipSTR attempts to assemble the sequences upstream and downstream of the STR (flank) to identify any potential SNPs it should consider. This assembly process fails if the resulting assembly graph contains a cycle, resulting in this filter |
| FLANK_ASSEMBLY_INDEL | This filter is triggered if the assembly process identifies an insertion or deletion in the flanks. These indels are problematic for HipSTR's model and thus it does not attempt to genotype the sample |
| FLANK_INDEL_FRAC | When genotyping is complete, HipSTR determines the maximum-likelihood alignment of each read relative to its sample's called alleles. If a large fraction of the resulting alignments have indels in the flanks, it's a strong indicator that they're misaligned and the sample's genotype is therefore ignored |
| LOW_FREQUENCY_ALT_FLANK | Flanking sequences identified by the assembly process in each sample are pooled together to generate all candidate haplotypes. As the number of haplotypes grows exponentially with the number of such sequences, HipSTR conserves time by discarding flanks that are only present in a few samples. If a sample's data supports a low-frequency flank, it is not genotyped. To adjust this frequency cutoff, use the --min-flank-freq option |
Although HipSTR mitigates many of the most common sources of STR genotyping errors, it's still extremely important to filter the resulting VCFs to discard low quality calls. To facilitate this process, the VCF output contains various FORMAT and INFO fields that are usually indicators of problematic calls. The INFO fields indicate the aggregate data for a locus and, if certain flags are raised, may suggest that the entire locus should be discarded. In contrast, FORMAT fields are available on a per-sample basis for each locus and, if certain flags are raised, suggest that some samples' genotypes should be discarded. The list below includes some of these fields and how they can be informative:
- DP: Reports the total depth/number of informative reads for all samples at the locus. The mean coverage per-sample can obtained by dividing this value by the number of samples with non-missing genotypes. In general, genotypes with a low mean coverage are unreliable because the reads may only have captured one of the two alleles if an individual is heterozygous.
- DSTUTTER: Reports the total number of reads at a locus with what HipSTR thinks is a stutter artifact. If the total fraction of reads with stutter (DSTUTTER/DP) is high, genotypes for a locus will be unreliable because the reads frequently don't reflect the true underlying genotype. A high fraction of stutter-containing reads can be caused by too much PCR amplification, a duplicated locus that is mapping to a single location in the genome, or a failure of HipSTR to identify sufficient candidate alleles.
- DFLANKINDEL: Reports the total number of reads for which the maximum likelihood alignment contains an indel in the regions flanking the STR. A high fraction of reads with this artifact (DFLANKINDEL/DP) can be caused by an actual indel in a region neighboring the STR. However, it can also arise if HipSTR fails to identify sufficient candidate alleles. When these alleles are very different in size from the candidate alleles or are non-unit multiples, they're frequently aligned as indels in the flanking sequences.
- Q: Reports the posterior probability of the genotype. We've found that this is the best indicator of quality of an individual sample's genotype and almost always use it to filter calls.
- DP, DSTUTTER and DFLANKINDEL: Identical to the INFO field case, these fields are also available for each sample and can be used in the same way to identify problematic individual calls.
- AB and FS: Quantify the log10 p-value of the allele bias and the Fisher strand bias, respectively. Large negative values indicate that the degree of bias observed is very unlikely to occur by random chance. In the case of AB, this indicates that the number of reads observed per allele is unlikely given the predicted genotype. In the case of FS, this indicates that there is a non-random association between sequencing strand and the allele each read is assigned to, suggesting that sequencing errors may be causing one of the reported alleles. Note that these fields are only applicable to diploid genotypes.
So what thresholds do we suggest for each of these fields? The answer really depends on the quality of the sequencing data, the ploidy of the chromosome and the downstream applications. However, we typically apply the following filters usings scripts we've provided in the scripts subdirectory of the HipSTR folder:
python scripts/filter_vcf.py --vcf diploid_calls.vcf.gz
--min-call-qual 0.9
--max-call-flank-indel 0.15
--max-call-stutter 0.15
--min-call-allele-bias -2
--min-call-strand-bias -2
python scripts/filter_haploid_vcf.py --vcf haploid_calls.vcf.gz
--min-call-qual 0.9
--max-call-flank-indel 0.15
--max-call-stutter 0.15
The resulting VCF, which is printed to the standard output stream, will omit calls on a sample-by-sample basis in which any of the following conditions are met: i) the posterior < 90%, ii) more than 15% of reads have a flank indel or iii) more than 15% of reads have a stutter artifact. For the diploid VCF, these filters will also remove genotypes with an allele bias or Fisher strand bias p-value less than 0.01 (10^-2). Calls for samples failing these criteria will be replaced with a "." missing symbol as per the VCF specification. For more filtering options, type either
python scripts/filter_vcf.py -h
python scripts/filter_haploid_vcf.py -h
| Option | Description |
|---|---|
| viz-out aln_viz.gz | Output a file of each locus' alignments for visualization with VizAln or VizAlnPdf Why? You want to visualize or inspect the STR genotypes |
| log log.txt | Output the log information to the provided file (Default = Standard error) |
| haploid-chrs list_of_chroms | Comma separated list of chromosomes to treat as haploid (Default = all diploid) Why? You're analyzing a haploid chromosome like chrY |
| no-rmdup | Don't remove PCR duplicates. By default, they'll be removed Why? Your sequencing data is for PCR-amplified regions |
| use-unpaired | Use unpaired reads when genotyping (Default = False) Why? Your sequencing data only contains single-ended reads |
| snp-vcf phased_snps.vcf.gz | Bgzipped input VCF file containing phased SNP genotypes for the samples to be genotyped. These SNPs will be used to physically phase STRs Why? You have available phased SNP genotypes |
| bam-samps list_of_read_groups | Comma separated list of samples in same order as BAM files. Assign each read the sample corresponding to its file. By default, each read must have an RG tag and and the sample is determined from the SM field Why? Your BAM file RG tags don't have an SM field |
| bam-libs list_of_read_groups | Comma separated list of libraries in same order as BAM files. Assign each read the library corresponding to its file. By default, each read must have an RG tag and and the library is determined from the LB field NOTE: This option is required when --bam-samps has been specified Why? Your BAM file RG tags don't have an LB tag |
| lib-field rg_field | Read group field used to assign each read a library. By default, the library is determined from the LB field associated with RG Why? Your BAM file RG tags don't have an LB tag and you want to use a different tag instead (e.g. SM) |
| def-stutter-model | For each locus, use a stutter model with PGEOM=0.9 and UP=DOWN=0.05 for in-frame artifacts and PGEOM=0.9 and UP=DOWN=0.01 for out-of-frame artifacts Why? You have too few samples for stutter estimation and don't have stutter models |
| min-reads num_reads | Minimum total reads required to genotype a locus (Default = 100) Why? Refer to the discussion above |
| output-filters | Write why individual calls were filtered to the VCF (Default = False) |
This list is comprised of the most useful and frequently used additional options, but is not all encompassing. For a complete list of options, please type
./HipSTR --help
When deciphering and inspecting STR calls, it's extremely useful to visualize the supporting reads. HipSTR facilitates this through the viz-out option, which writes a compressed file containing alignments for each call that can be readily visualized using the VizAln command included in HipSTR's main directory. If you're interested in visualizing alignments, you first need to index the file using tabix.
For example, if you ran HipSTR with the option --viz-out aln.viz.gz, you should use the command
tabix -p bed aln.viz.gz
to generate a tabix index for the file so that we can rapidly extract alignments for a locus of interest. This command only needs to be run once after the file has been generated.
You could then visualize the calls for sample NA12878 at locus chr1 3784267 using the command
./VizAln aln.viz.gz chr1 3784267 NA12878
This command will automatically open a rendering of the alignments in your browser and might look something like:
 The top bar represents the reference sequence and the red text indicates the name of the sample and its associated call at the locus. The remaining rows indicate the alignment for each read used in genotyping. In this particular example, 14 reads have an 8bp deletion and 14 reads have a 4bp insertion. HipSTR therefore genotypes this sample as -8 | 4
The top bar represents the reference sequence and the red text indicates the name of the sample and its associated call at the locus. The remaining rows indicate the alignment for each read used in genotyping. In this particular example, 14 reads have an 8bp deletion and 14 reads have a 4bp insertion. HipSTR therefore genotypes this sample as -8 | 4
If we wanted to inspect all calls for the same locus, we could use the command
./VizAln aln.viz.gz chr1 3784267
To facilitate rendering these images for publications, we've also created a similar script that converts these alignments into a PDF. This script can only be applied to one sample at a time, but the image above can be generated in a file alignments.pdf as follows:
./VizAlnPdf aln.viz.gz chr1 3784267 NA12878 alignments 1
NOTE: Because the viz-out file can become fairly large if you're genotyping thousands of loci or thousands of samples, in some scenarios it may be best to rerun HipSTR using this option on the subset of loci which you wish to visualize.
HipSTR requires BAM/CRAM files produced by any indel-sensitive aligner. These files must have been sorted by position using the samtools sort command and then indexed using samtools index. To associate a read with its sample of interest, HipSTR uses read group information in the BAM/CRAM header lines. These *@*RG lines must contain an ID field, an LB field indicating the library and an SM field indicating the sample. For example, if a BAM/CRAM contained the following header line
@RG ID:RUN1 LB:ERR12345 SM:SAMPLE789
an alignment with the RG tag
RG:Z:RUN1
will be associated with sample SAMPLE789 and library ERR12345. In this manner, HipSTR can analyze BAMs/CRAMs containing more than one sample and/or more than one library and can handle cases in which a single sample's reads are spread across multiple files.
Alternatively, if your BAM/CRAM files lack RG information, you can use the bam-samps and bam-libs flags to specify the sample and library associated with each file. In this setting, however, a BAM/CRAM can only contain a single library and a single read group. For example, the command
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.cram
--fasta genome.fa
--regions str_regions.bed
--str-vcf str_calls.vcf.gz
--bam-samps SAMPLE1,SAMPLE1,SAMPLE2,SAMPLE3
--bam-libs LIB1,LIB2,LIB3,LIB4
essentially tells HipSTR to associate all the reads in the first two BAMS with SAMPLE1, all the reads in the third file with SAMPLE2 and all the reads in the last BAM with SAMPLE3.
HipSTR can analyze both BAM and CRAM files simultaneously, so if your project contains a mixture of these two file types, HipSTR will automatically perform CRAM decompression as necessary. When analyzing CRAM files, please ensure that the file provided to --fasta is the same FASTA file used during CRAM generation. Otherwise, CRAM decompression will likely fail and bizarre behavior may occur.
The BED file containing each STR region of interest is a tab-delimited file comprised of 5 required columns and one optional column:
- The name of the chromosome on which the STR is located
- The start position of the STR on its chromosome
- The end position of the STR on its chromosome
- The motif length (i.e. the number of bases in the repeat unit)
- The number of copies of the repeat unit in the reference allele
The 6th column is optional and contains the name of the STR locus, which will be written to the ID column in the VCF. Below is an example file which contains 5 STR loci
NOTE: The table header is for descriptive purposes. The BED file should not have a header
| CHROM | START | END | MOTIF_LEN | NUM_COPIES | NAME |
|---|---|---|---|---|---|
| chr1 | 13784267 | 13784306 | 4 | 10 | GATA27E01 |
| chr1 | 18789523 | 18789555 | 3 | 11 | ATA008 |
| chr2 | 32079410 | 32079469 | 4 | 15 | AGAT117 |
| chr17 | 38994441 | 38994492 | 4 | 12 | GATA25A04 |
| chr17 | 55299940 | 55299992 | 4 | 13 | AAT245 |
We've provided various BED files containing STR loci for different organisms, including humans, here
For other model organisms, we recommend that you modify the framework we used to build the mouse BED file.
For more information on the VCF file format, please see the VCF spec.
INFO fields contains aggregated statistics about each genotyped STR in the VCF. The INFO fields reported by HipSTR primarily describe the learned/supplied stutter model for the locus, the STR's reference coordinates (START and END) and information about the allele counts (AC) and number of reads used to genotype all samples (DP).
| FIELD | DESCRIPTION |
|---|---|
| INFRAME_PGEOM | Parameter for in-frame geometric step size distribution |
| INFRAME_UP | Probability that stutter causes an in-frame increase in obs. STR size |
| INFRAME_DOWN | Probability that stutter causes an in-frame decrease in obs. STR size |
| OUTFRAME_PGEOM | Parameter for out-of-frame geometric step size distribution |
| OUTFRAME_UP | Probability that stutter causes an out-of-frame increase in obs. STR size |
| OUTFRAME_DOWN | Probability that stutter causes an out-of-frame decrease in obs. STR size |
| BPDIFFS | Base pair difference of each alternate allele from the reference allele |
| START | Inclusive start coodinate for the repetitive portion of the reference allele |
| END | Inclusive end coordinate for the repetitive portion of the reference allele |
| PERIOD | Length of STR motif |
| AN | Total number of alleles in called genotypes |
| REFAC | Reference allele count |
| AC | Alternate allele counts |
| NSKIP | Number of samples not genotyped due to various issues |
| NFILT | Number of samples that were originally genotyped but have since been filtered |
| DP | Total number of reads used to genotype all samples |
| DSNP | Total number of reads with SNP information |
| DSTUTTER | Total number of reads with a stutter indel in the STR region |
| DFLANKINDEL | Total number of reads with an indel in the regions flanking the STR |
FORMAT fields contain information about the genotype for each sample at the locus. In addition to the most probable phased genotype (GT), HipSTR reports information about the posterior likelihood of this genotype (PQ) and its unphased analog (Q). Other useful information reported are the number of reads that were used to determine the genotype (DP) and whether these had any alignment artifacts (DSTUTTER and DFLANKINDEL).
| FIELD | DESCRIPTION |
|---|---|
| GT | Genotype |
| GB | Base pair differences of genotype from reference |
| Q | Posterior probability of unphased genotype |
| PQ | Posterior probability of phased genotype |
| DP | Number of valid reads used for sample's genotype |
| DSNP | Number of reads with SNP phasing information |
| PDP | Fractional reads supporting each haploid genotype |
| GLDIFF | Difference in likelihood between the reported and next best genotypes |
| DSNP | Total number of reads with SNP information |
| PSNP | Number of reads with SNPs supporting each haploid genotype |
| DSTUTTER | Number of reads with a stutter indel in the STR region |
| DFLANKINDEL | Number of reads with an indel in the regions flanking the STR |
| AB | log10 of the allele bias pvalue, where 0 is no bias and more negative values are increasingly biased. For homozygous genotypes, this can be negative if the haplotypes are heterozygous |
| FS | log10 of the strand bias pvalue from Fisher's exact test, where 0 is no bias and more negative values are increasingly biased. For homozygous genotypes, this can be negative if the haplotypes are heterozygous |
| DAB | Number of reads used in the allele bias calculation |
| ALLREADS | Base pair difference observed in each read's Needleman-Wunsch alignment |
| MALLREADS | Maximum likelihood bp diff in each read based on haplotype alignments |
| GL | log-10 genotype likelihoods |
| PL | Phred-scaled genotype likelihoods |
To model PCR stutter artifacts, we assume that there are three types of stutter artifacts:
- In-frame changes: Change the size of the STR in the read by multiples of the repeat unit. For instance, if the repeat motif is AGAT, in-frame changes could lead to differences of -8, -4, 4, 8, and so on.
- Out-of-frame changes: Change the size of the STR by non-multiples of the repeat unit. For instance, if the repeat motif is AGAT, out-of-frame changes could lead to differences of -3, -2, -1, 1, 2, 3 and so on.
- No stutter change: The size of the STR in the read is the same as the size of the underlying STR.
Stutter model files contain the information necessary to model each of these artifacts in a tab-delimited BED-like format with exactly 9 columns. An example of such a file is as follows:
| CHROM | START | END | IGEOM | IDOWN | IUP | OGEOM | ODOWN | OUP | PERIOD |
|---|---|---|---|---|---|---|---|---|---|
| chr1 | 18789523 | 18789555 | 0.8 | 0.01 | 0.05 | 0.9 | 0.001 | 0.001 | 3 |
| chr2 | 32079410 | 32079469 | 0.9 | 0.01 | 0.01 | 0.9 | 0.001 | 0.001 | 4 |
| chr17 | 38994441 | 38994492 | 0.9 | 0.001 | 0.001 | 0.9 | 0.001 | 0.001 | 4 |
| chr9 | 3859351 | 3859400 | 0.9 | 0.1 | 0.02 | 0.9 | 0.001 | 0.001 | 2 |
NOTE: The table header is for descriptive purposes. The stutter file should not have a header
Each of the stutter parameters is defined as follows:
| VARIABLE | DESCRIPTION |
|---|---|
| IDOWN | Probability that in-frame changes decrease the size of the observed STR allele |
| IUP | Probability that in-frame changes increase the size of the observed STR allele |
| ODOWN | Probability that out-of-frame changes decrease the size of the observed STR allele |
| OUP | Probability that out-of-frame changes increase the size of the observed STR allele |
| IGEOM | Parameter governing geometric step size distribution for in-frame changes |
| OGEOM | Paramter governing geometric step size distribution for out-of-frame changes |
| PERIOD | Length of STR motif |
- Can I run HipSTR if my dataset only contains single-ended reads?
Yes. HipSTR is designed for paired-end reads and uses mate pair information to filter reads that are potentially aligned to an incorrect STR prior to genotyping. By default, HipSTR therefore removes all reads without mate pairs. However, if your dataset only contains single-ended reads, specify the use-unpaired option to avoid performing this filtering. - Can I use HipSTR to analyze PCR-amplified reads?
Yes. As HipSTR was designed to analyze WGS data, HipSTR automatically identifies and filters out PCR duplicates prior to genotyping. When analyzing PCR-amplified reads, HipSTR will label most reads as PCR duplicates as they share exactly the same coordinates. To overcome this issue, specify the no-rmdup option to disable duplicate removal when analyzing this type of data. - Why are some of the STRs in my BED file not present in the output VCF?
HipSTR only genotypes a region if at least min-reads and at most max-reads overlap the STR. It then attempts to learn the stutter model (if appropriate), build haplotypes for the region and perform genotyping. If any of these stages is unsuccessful, it skips the STR and continues on to the next region. The log file contains the failure reason for each failed region as well as an overall summary of why regions were skipped at the end of the log. - How can I run HipSTR if I have too few samples to learn stutter models and don't have external ones?
In this scenario, you can run HipSTR with def-stutter-model. Invoking this option will disable the algorithm it uses to learn stutter models. Instead, HipSTR will use the same fixed stutter model to genotype every locus. We don't recommend using this option unless necessary, as genotypes are more accurate if you learn a specific model for each STR. - What sequencing platforms does HipSTR support? HipSTR was designed to analyze Illumina sequencing data. We do not recommend running it on PacBio or Oxford Nanopore data, as the difference in error profiles will be problematic
If you're having trouble getting your analysis up and running:
i. Check out the HipSTR tutorial at https://hipstr-tool.github.io/HipSTR-tutorial
ii. Type ./HipSTR --help for details about each command line option
iii. Email us at [email protected]
If you encounter a bug/issue or have a feature request:
i. File an issue on GitHub (https://github.com/tfwillems/HipSTR)
ii. Email us at [email protected]
If you found HipSTR useful, we would appreciate it if you could cite our manuscript describing HipSTR and its applications: Genome-wide profiling of heritable and de novo STR variations